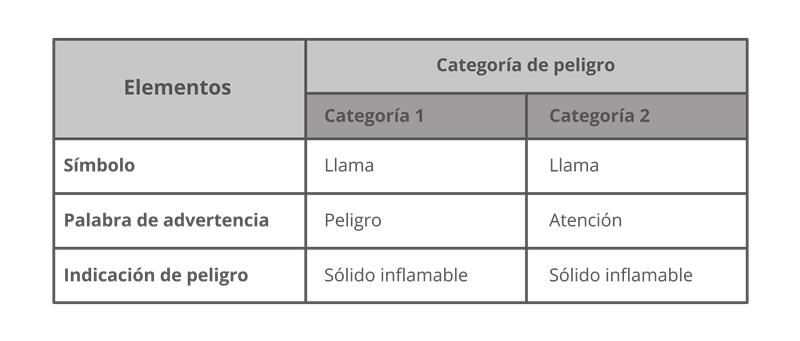
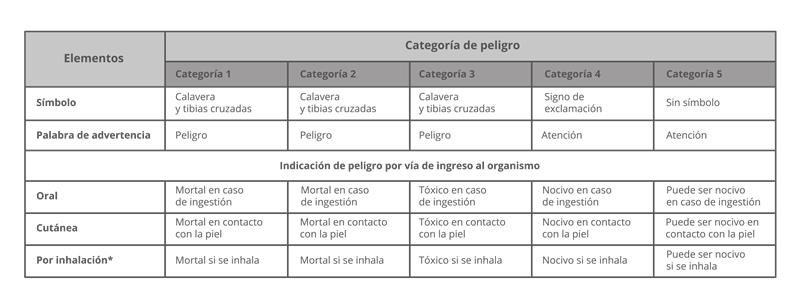
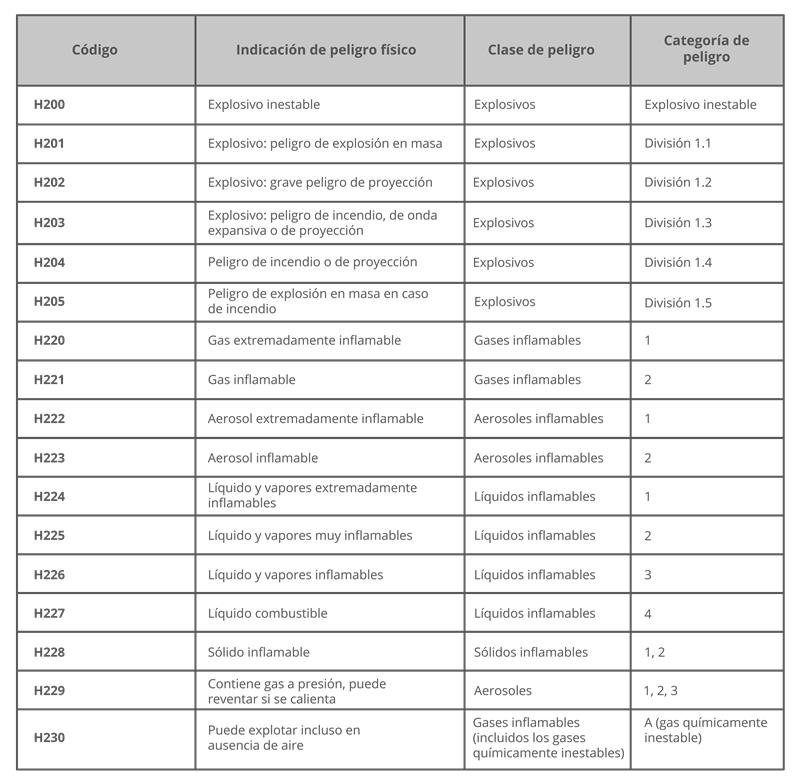
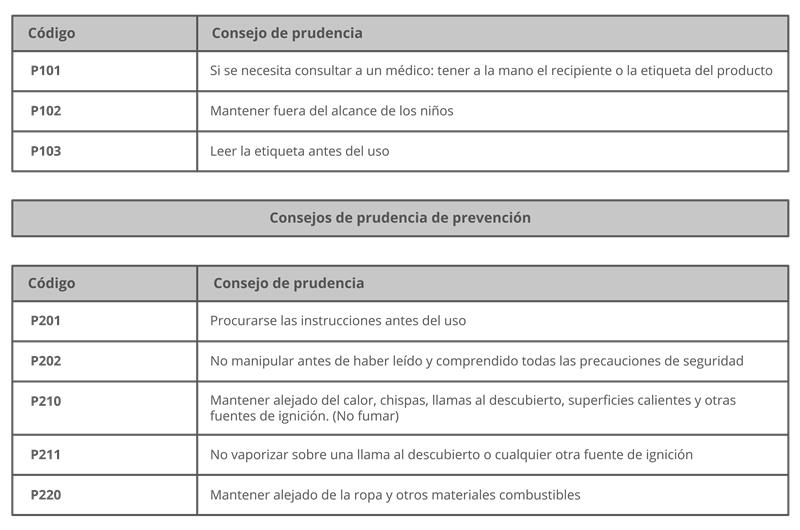
ISO 45001:2018
La norma ISO 45001:2018 que se publicó en marzo del presente año sustituirá a OHSAS 18001:2007, para ello se estableció un periodo de transición de 3 años a partir de la fecha de publicación de la norma. Las organizaciones que actualmente tienen una certificación en OHSAS 18001 deberán migrar a ISO 45001 antes de marzo de 2021.
Global Standards alienta a sus clientes certificados en OHSAS 18001 a ejecutar su evento de transición mediante una auditoría de seguimiento, recertificación o una auditoría especial, a más tardar el 31 de septiembre del 2020. Pueden consultar información con mayor detalle en el comunicado número 27, publicado en el sitio web de Global Standards.
ISO 22000:2018
La nueva norma ISO 22000:2018 fue publicada en junio del año en curso, conforme a los acuerdos de los miembros del Foro Internacional de Acreditación se estipuló un periodo de 3 años para la transición de las organizaciones certificadas en la versión anterior. Por lo tanto, todos los certificados en la versión 2005 perderán su validez en junio del 2021, y las organizaciones certificadas deberán realizar su auditoría de transición antes de esa fecha.
Esperamos la actualización de la regla de acreditación 22 de ANAB, nuestro organismo de acreditación, para ofrecer más información sobre el proceso de esta transición.
FSSC 22000 versión 5
La Fundación FSSC 22000 anunció la actualización de su programa 4.1 a la versión 5, que en general busca alinearse a los requisitos de la nueva versión de ISO 22000:2018. La nueva versión de FSSC 22000 incrementará sus alcances.
Se espera que la versión 5 se publique en mayo del próximo año y el plazo de transición para las organizaciones certificadas en FSSC 2200 será hasta junio de 2021, alineándose al periodo de transición de ISO 22000:2018.
PrimusGFS versión 2.1-2c se actualizó
La versión 3 de PrimusGFS fue reconocida por la Iniciativa Global de Seguridad Alimentaria, por lo que el dueño del programa PrimusGFS publicó oficialmente la nueva versión del programa en agosto de este año; el proceso de transición es simple, PrimusGFS acepta auditorías en la versión 2.1-2c y en la versión 3, desde agosto hasta diciembre de 2018, siendo obligatorio que a partir de enero de 2019 todas las auditorías se ejecuten en la versión 3.
Por lo tanto, las organizaciones certificadas actualmente en la versión 2.1-2c deberán ejecutar su evento de recertificación en la nueva versión el próximo año.
Código SQF
El Código SQF también será actualizado, aún no tenemos más información, pero es importante adelantar que la versión 8 sufrirá cambios significativos que se verán reflejados en una nueva versión del código.